
New Protein Design Methods for De Novo Small Molecule Binding Sites
Published in PLoS Computational Biology, 2020
Recommended citation: Lucas JE, Kortemme T (2020) New computational protein design methods for de novo small molecule binding sites. PLOS Computational Biology 16(10): e1008178. https://doi.org/10.1371/journal.pcbi.1008178 https://doi.org/10.1371/journal.pcbi.1008178
Protein binding to small molecules is fundamental to many biological processes, yet current state-of-the-art methods are unable to predictively design this functionality de novo. In lieu of existing design methods that rely on existing binding site definitions or protein scaffolds with existing shape complementarity for a target ligand, we introduce new methods that utilize pools of discrete protein interactions with defined chemical moieties observed in the Protein Data Bank. We use the Rosetta Molecular Modeling Suite to recombine residues in a contact pool to generate hundreds of thousands of energetically favorable binding sites for a target ligand. These composite binding sites are built into existing scaffold proteins with high accuracy to impart desired functionality. In addition, we apply observed protein-fragment interactions to augment Rosetta’s conventional design machinery and improve key design metrics responsible for predicting design success. We demonstrate that our method reliably builds diverse binding sites into various scaffold proteins for several target molecules. Our generalizable de novo ligand binding site design method will lay the foundation for versatile design of protein to interface previously unattainable molecules for applications in medical diagnostics and synthetic biology.
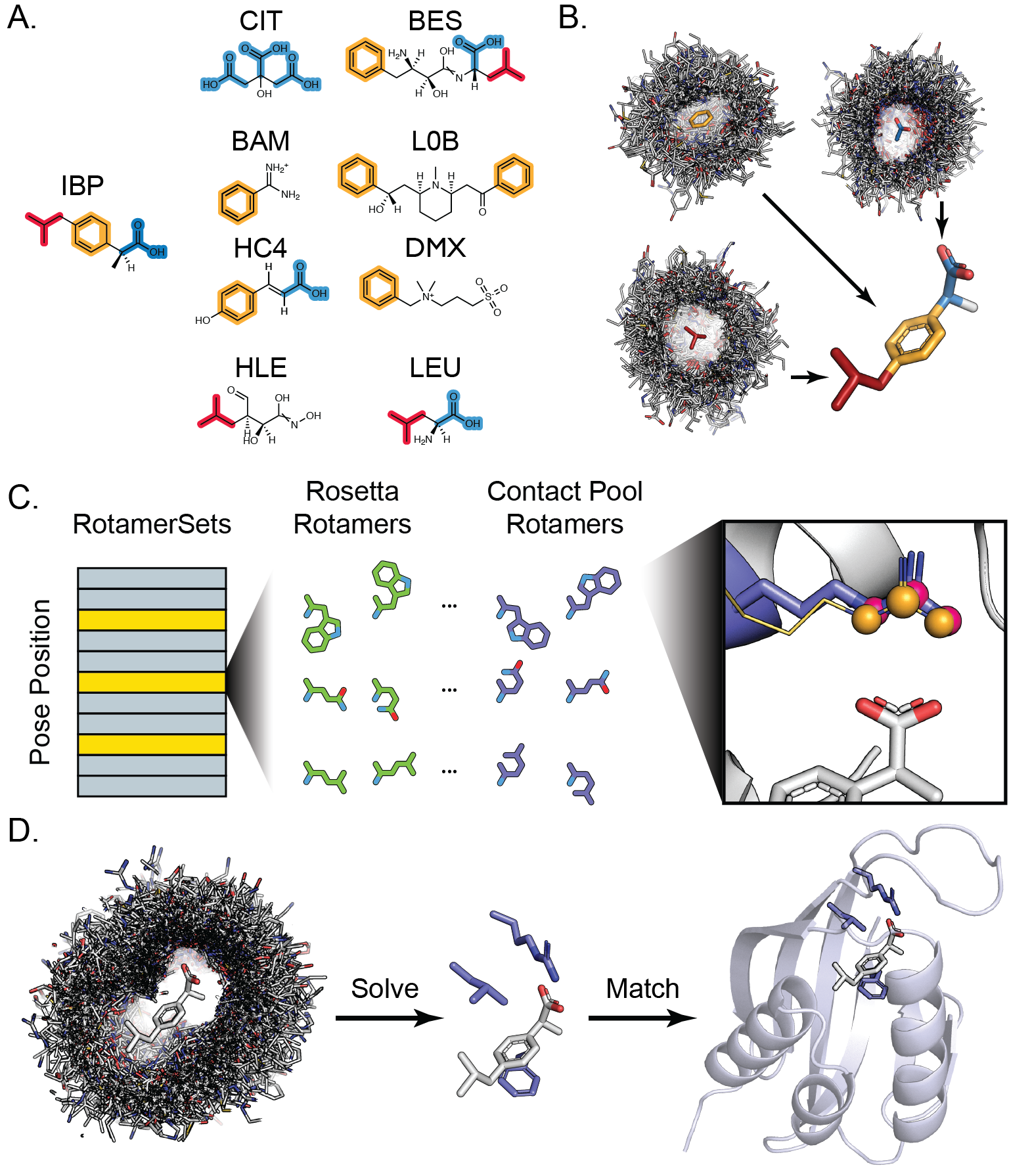
A. Target small molecules such as ibuprofen can be decomposed into distinct chemical moieties (highlighted red, orange, and blue), or fragments, that are present as a substructure in a wide range of molecules bound to proteins in the PDB. B. Protein complexes bound to substructure-containing molecules are systematically parsed to generate contact pools representing all contact modes present in the PDB with each fragment. These contacts are mapped onto the full target ligand to create a conformer-specific contact pool for downstream steps. C. Contact pools are used to supplement the set of rotamers generated by Rosetta’s Packer during design. Rotamers are built in the context of a potential ligand interface and any rotamers (purple sticks, pink spheres) that recapitulate an interaction in the contact pool (gold lines, orange spheres) is added to the Packer RotamerSet and provided a score bonus with the special_rot score term. D. A simulated annealing protocol is used to assemble hundreds of thousands of three-residue composite binding sites from a target ligand contact pool. RosettaMatch is then used to find scaffold proteins that can geometrically accommodate a composite binding site solution.